DNA methylation is a pivotal mechanism regulating gene expression, characterized by the covalent attachment of a methyl group to the 5th carbon of cytosine at specific CpG dinucleotide sites mediated by DNA methyltransferases. This process plays a crucial role in DNA replication and transcriptional regulation, encompassing de novo methylation and maintenance methylation pathways. De novo methylation entails the methylation of previously unmethylated DNA strands catalyzed by methyltransferases, whereas maintenance methylation occurs when one strand of the DNA duplex is already methylated, leading to methylation of the unmethylated strand. These methylation modalities collectively maintain the stability and dynamic equilibrium of the methylome.
Principles of DNA Methylation
DNA methylation represents a extensively researched and longstanding epigenetic regulatory mechanism. Broadly, it refers to the chemical modification process where specific bases within the DNA sequence, under the catalytic action of DNA methyltransferases utilizing S-adenosyl methionine as the methyl donor, undergo covalent attachment of methyl groups. This modification can occur at various sites such as the C-5 position of cytosine, N-6 position of adenine, and G-7 position of guanine. In most contexts, DNA methylation predominantly refers to the methylation process of the 5th carbon of cytosine in CpG dinucleotides. The resultant product, 5-methylcytosine, represents the primary form of DNA methylation in eukaryotes, including plants and animals, and constitutes the sole known form of DNA methylation in mammals. As a relatively stable modification state, DNA methylation can be inherited to offspring DNA during DNA replication via the action of DNA methyltransferases, thus constituting a crucial epigenetic mechanism.
Classification of DNA Methyltransferases
Within the genome, the establishment of DNA methylation patterns relies on the activity of DNA methyltransferases. These enzymes encompass maintenance DNA methyltransferases (also known as Dnmt1 or maintenance methyltransferases) and de novo methyltransferases. Based on sequence homology and function, eukaryotic DNA methyltransferases further subdivide into four classes: Dnmt1/MET1, Dnmt2, CMTs, and Dnmt3. Dnmt1/MET1 enzymes are responsible for maintaining the methylation status of CG sequences. CMTs enzymes are exclusively found in plants, characterized by their catalytic domains T and IV being buried within the chromosomal major area and their specific maintenance of CG sequence methylation. Dnmt3 enzymes have been identified in mice, humans, and zebrafish. Among these, Dnmt3a and Dnmt3b exhibit higher expression levels in undifferentiated embryonic stem cells but lower expression levels in somatic cells. Their primary function is de novo methylation, although they also contribute to maintenance methylation and are responsible for methylation of repetitive sequences.
Detection method of DNA methylation
1) Bisulfite Sequencing
Bisulfite sequencing, a widely employed method for DNA methylation analysis, entails the conversion of unmethylated cytosines to uracils via specific chemical reactions, generating uracil bands in the DNA sequence that are easily discernible. Methylated cytosines remain unaffected, retaining their original state. Subsequently, DNA fragments treated with bisulfite are selectively amplified through PCR and their sequence information is acquired using DNA sequencing technology. By aligning the sequencing results with the original sequence, one can precisely enumerate the number and distribution of methylated sites, thereby assessing the degree of DNA methylation. This method, renowned for its high sensitivity and robust reproducibility, is particularly suited for detecting methylation levels in individual genes or a limited set thereof.
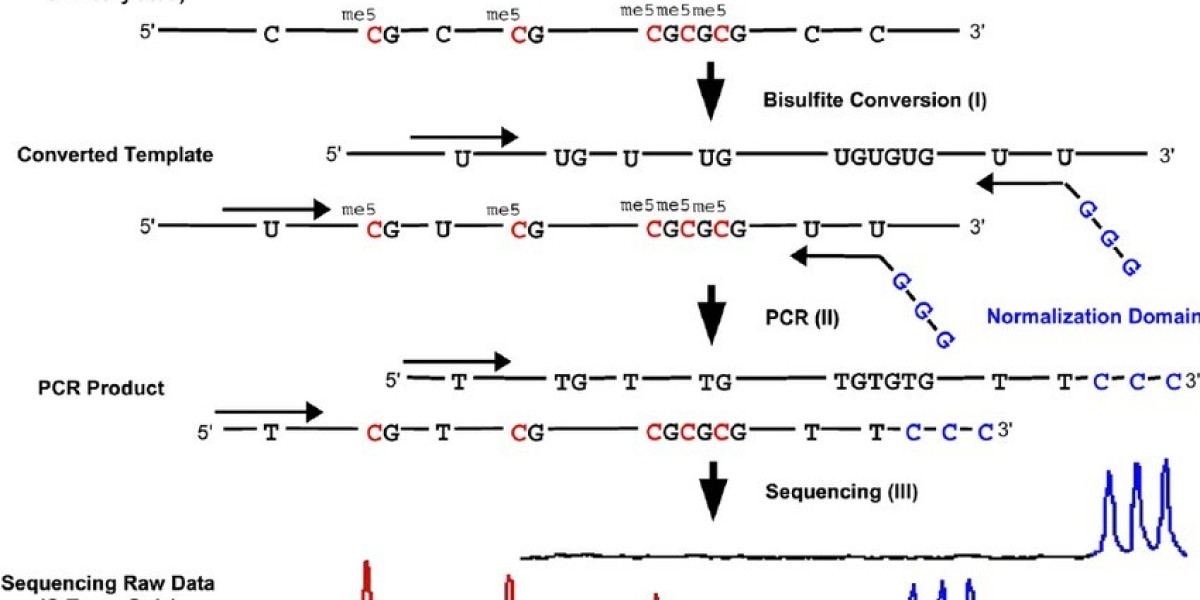
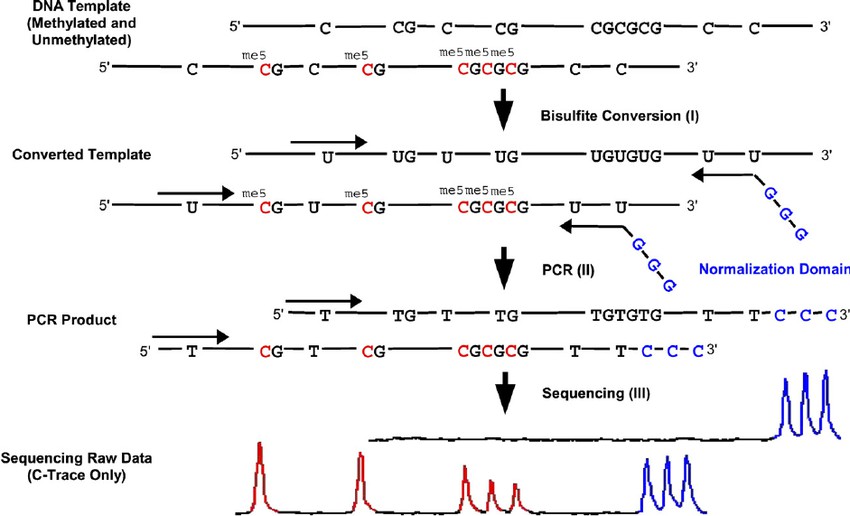 Principle of the quantitative bisulfite sequencing method (Dimo Dietrich et al,.2019 )
Principle of the quantitative bisulfite sequencing method (Dimo Dietrich et al,.2019 )
2) RRBS
Reduced Representation Bisulfite Sequencing (RRBS) utilizes bisulfite treatment and restriction enzyme digestion to selectively amplify DNA fragments enriched with high methylation content. This method expedites the accurate detection of heavily methylated CpG sites within the genome.
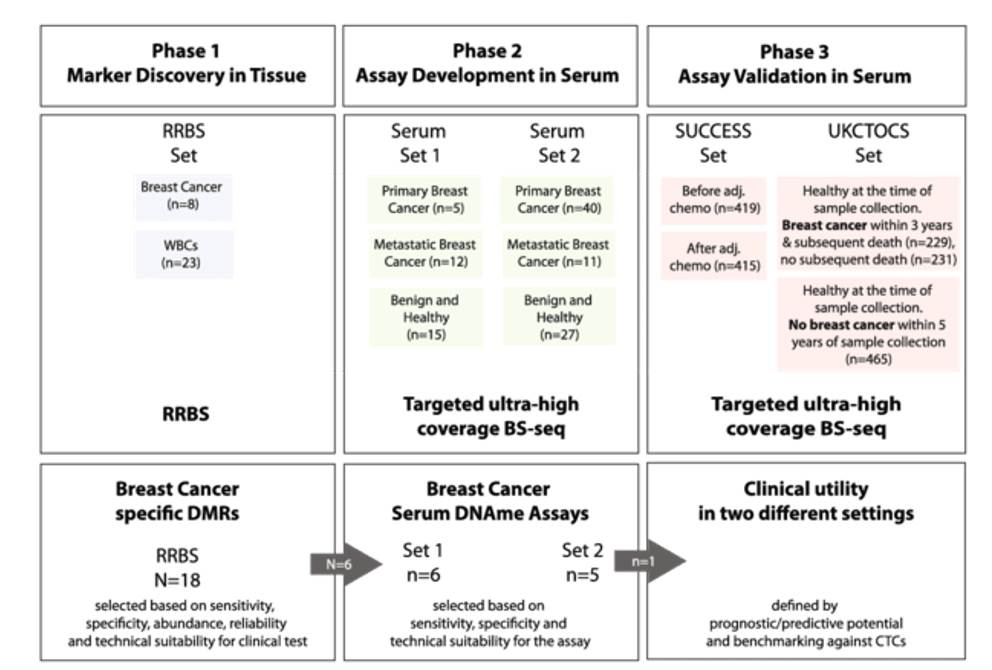 Study design of Reduced representation bisulfite sequencing (RRBS) (Martin Widschwendter et al,. 2017)
Study design of Reduced representation bisulfite sequencing (RRBS) (Martin Widschwendter et al,. 2017)
3) WGBS
Whole Genome Bisulfite Sequencing (WGBS) represents a high-throughput approach capable of comprehensively assessing the methylation levels of every individual cytosine base throughout the entire genome. Achieving precise methylation data necessitates deep sequencing coverage with this method.
 Whole-Genome Bisulfite Sequencing of DNA Methylomes of Mouse Embryonic Stem Cells (Ehsan Habibi et al,. 2013)
Whole-Genome Bisulfite Sequencing of DNA Methylomes of Mouse Embryonic Stem Cells (Ehsan Habibi et al,. 2013)
4) MeDIP-seq
Methylated DNA Immunoprecipitation Sequencing (MeDIP-seq) leverages immunoprecipitation with antibodies targeting 5-methylcytosine, succeeded by high-throughput sequencing. This methodology facilitates the comprehensive detection of methylation status across all CpG sites within the genome.
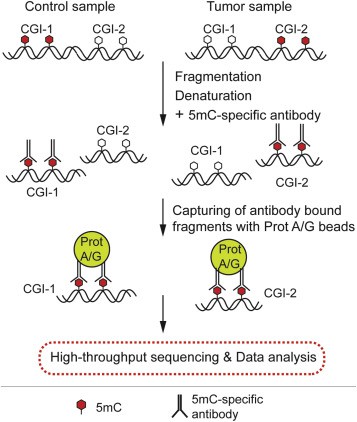 Methylated DNA immunoprecipitation sequencing (MeDIP-seq). (Tibor A. Rauch, Gerd P. Pfeifer, 2019
Methylated DNA immunoprecipitation sequencing (MeDIP-seq). (Tibor A. Rauch, Gerd P. Pfeifer, 2019
For more information about DNA Methylation Sequencing Methods, refer to "Overview of DNA Methylation Sequencing Methods".
Applications of DNA methylation
Disease Pathogenesis
Within the realm of oncology, alterations in the methylation patterns of key tumor-associated genes orchestrate a cascade of events culminating in gene silencing or overexpression, thereby propelling tumorigenesis. Exemplifying this, in breast cancer, frequent DNA methylation events targeting tumor suppressor genes including BRCA1 and TP53 engender diminished or abrogated gene expression, thereby heightening susceptibility to malignancy. Furthermore, deviations in DNA methylation levels intricately correlate with tumor aggressiveness and prognostic outcomes. Investigations underscore that tissues exhibiting heightened methylation profiles typically manifest inferior survival rates and augmented metastatic propensities relative to counterparts evincing lower methylation levels. Consequently, DNA methylation emerges as a pivotal entity in the landscape of early cancer detection and diagnosis.
Cardiovascular Diseases: Beyond oncology, the perturbation of methylated gene expression and the modulation of DNA methylation levels intricately contribute to the etiology of cardiovascular maladies encompassing hypertension, coronary artery disease, and myocardial infarction. Additionally, in the realm of inherited disorders, DNA methylation serves as a diagnostic arbiter for select monogenic conditions such as cystic fibrosis and hemophilia.
Expanding the purview, emerging evidence implicates DNA methylation in the pathogenesis of psychiatric disorders, including autism spectrum disorder and schizophrenia. Furthermore, investigations suggest a potential nexus between DNA methylation patterns and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus, underscoring its multifaceted role in disease pathophysiology.
Exploring the Landscape of Disease Biomarkers
Within the realm of cancer research, DNA methylation emerges as a cornerstone, delineating distinct methylation signatures across diverse malignancies. Interrogating the DNA methylation landscape of neoplastic tissues facilitates early detection, prognostic evaluation, and recurrence surveillance. Noteworthy is the prognostic relevance of gene-specific methylation statuses in lung cancer, colorectal cancer, breast cancer, and other tumor types, elucidating survival probabilities and recurrence propensities.
In the domain of genetic diseases, DNA methylation assumes a diagnostic mantle. By scrutinizing the methylation profiles of select genes, the identification and screening of monogenic inherited disorders, exemplified by cystic fibrosis and hemophilia, are streamlined.
Insights into the etiology of psychiatric disorders such as autism spectrum disorder and schizophrenia intertwine with the intricate landscape of DNA methylation. Analysis of the methylation status in individuals afflicted with psychiatric maladies not only deepens our understanding of disease pathogenesis but also furnishes novel avenues for diagnostic modalities and therapeutic interventions.
Expanding the purview to autoimmune diseases like rheumatoid arthritis and systemic lupus erythematosus, burgeoning research underscores the contributory role of DNA methylation. Evaluation of DNA methylation patterns among patients afflicted with autoimmune disorders augments our comprehension of disease mechanisms and unveils promising therapeutic strategies.
Breeding of Animals and Plants
In the realm of animal and plant breeding, research endeavors delve into the intricate interplay between methylation patterns and various phenotypic manifestations including growth, development, disease resistance, reproductive prowess, embryonic maturation, and senescence. This fundamental exploration serves as a linchpin in understanding the genetic underpinnings of desired traits in both animal and plant species.
Harnessing DNA methylation as a molecular beacon furnishes breeders with a potent tool for discerning individuals harboring coveted traits such as disease resilience, pest immunity, and heightened productivity among animal and plant populations. This molecular marker augments precision breeding efforts aimed at enhancing agricultural productivity and sustainability.
Moreover, the advent of genome editing technologies revolutionizes breeding paradigms by facilitating the targeted modulation of methylation profiles within animal and plant genomes. This molecular manipulation engenders precise control over gene expression dynamics, thereby augmenting the manifestation of desired phenotypic traits in progeny.
Environment
Delving into environmental considerations, the utilization of plants for remediation purposes emerges as a promising avenue. Certain botanical species exhibit a remarkable propensity for pollutant sequestration. By orchestrating alterations in the methylation status of these plants, their capacity for pollutant absorption and detoxification can be bolstered, thereby amplifying their efficacy in environmental cleanup endeavors.
Microbial degradation stands as a cornerstone in the arsenal of environmental remediation strategies. Manipulating the methylation profiles of microbial consortia holds promise for enhancing their enzymatic prowess in metabolizing organic pollutants, thereby facilitating the bioremediation of contaminated ecosystems.
Furthermore, molecular ecological inquiries harness the power of DNA methylation to unravel the intricacies of microbial community dynamics in polluted habitats. These investigations elucidate the structural and functional facets of microbial populations, shedding light on their roles in pollutant degradation processes and providing pivotal insights for the design of targeted pollution mitigation strategies.











